In all of our experiments the ions are cooled (either in a supersonic expansion or a cryogenic ion trap), which results in very well-defined structures that we characterize with IR spectroscopy. During this rapid cooling process, the ions can be trapped in many different local minima across the potential energy surface resulting in the presence of multiple conformational isomers. These isomers all contribute to the IR spectrum and render their analysis difficult. To overcome this problem, we have developed a pump-probe double resonance scheme that utilizes two tunable infrared lasers and either two or three stages of mass selection. First, a pump laser, whose frequency is scanned through the IR range, is intersected with the ion packet and followed by a mass-selection stage. When the frequency of the pump laser is resonant with and IR transition, it depletes the total tagged-ion ensemble of one particular isomer. The remaining tagged ions are then interrogated by a probe laser, whose frequency fixed to a particular infrared transition, and the photofragments are monitored after another stage of mass-selection. When both the pump and the probe lasers are tuned to transitions belonging to the same isomers, a depletion of the probe photofragmentation signal is produced, thus effectively generating the IR spectrum of a single isomer within the ensemble.
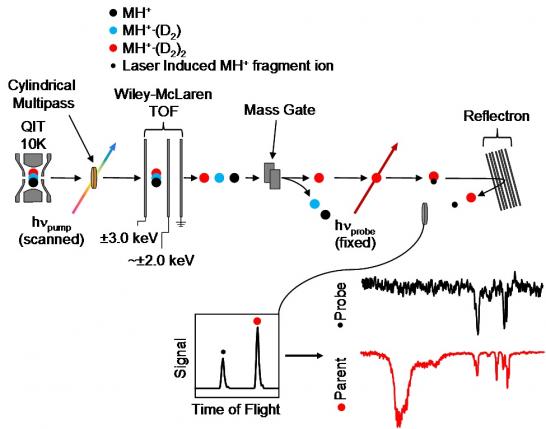
A schematic of this process is indicated above for ions cooled and tagged in a cryogenic ion trap and where the pump laser interacts with the ion packet directly after the trap and prior to the first mass-selection stage. A similar approach can also be used for Ar-tagged ions prepared in a supersonic expansion. In this arrangement, an additional mass-selection stage is used between the ion source and the pump-laser interaction. We have also use variations of this two lasers scheme to obtain isomer specific photoelectron spectra or to study the result of photo-induced reactions.
(1) Johnson, M. A.; Rostas, J.; Zare R. N. Optical-optical double resonance on cooled molecular ions: rotational assignments in the perturbed CO+2![]() -
-![]() system Chem. Phys. Lett., 92, 225-231 (1982).
system Chem. Phys. Lett., 92, 225-231 (1982).
(2) Elliott, B. M.; Relph, R. A.; Roscioli, J. R.; Bopp, J. C.; Gardenier, G. H.; Guasco, T. L.; Johnson M. A. Isolating the spectra of cluster ion isomers using Ar-“tag” -mediated IR-IR double resonance within the vibrational manifolds: Application to NO2−⋅H2O J. Phys. Chem. 129, 094303 (2008).
(3) Roscioli, J. R.; Hammer, N. I.; Johnson, M. A.; Diri, K.; Jordan, K. D. Exploring the correlation between network structure and electron binding energy in the (H2O)7− cluster through isomer-photoselected vibrational predissociation spectroscopy and ab initio calculations: Addressing complexity beyond types I-III J. Chem. Phys. 128, 104314 (2008).
(4) Relph, R. A.; Elliott, B. M.; Weddle, G. H.; Johnson, M. A.; Ding, J.; Jordan, K. D. Vibrationally Induced Interconversion of H-Bonded NO2−·H2O Isomers within NO2−·H2O·Arm Clusters Using IR−IR Pump−Probe through the OH and NO Stretching Vibrations J. Phys. Chem. A. 113, 975-981 (2009).
(5) Guasco, T. L.; Elliott, B. M.; Johnson, M. A., Ding, J.; Jordan, K. D. Isolating the Spectral Signatures of Individual Sites in Water Networks Using Vibrational Double-Resonance Spectroscopy of Cluster Isotopomers J. Phys. Chem. Lett. 1, 2396-2401 (2010).
(6) Leavitt, C. M.; Wolk, A. B.; Fournier, J. A.; Kamrath, M. Z.; Garand, E.; Van Stipdonk, M. J.; Johnson, M. A. Isomer-Specific IR–IR Double Resonance Spectroscopy of D2-Tagged Protonated Dipeptides Prepared in a Cryogenic Ion Trap J. Phys. Chem. Lett. 3, 1099-1105 (2012).